Mission
The research in Professor Zagorski's group focuses on unraveling the molecular mechanisms associated with protein misfolding and amyloid formation. Numerous analytical techniques are utilized, including nuclear magnetic resonance (NMR), circular dichroism, atomic force microscopy, electron microscopy, analytical ultracentrifugation, peptide synthesis and peptide purification using high performance liquid chromatography. Frequent collaborations with scientists in the School of Medicine are fundamental to the long-term goals of the group's research, which is the eradication of human disease. Current research efforts focus on proteins with important pathological features in neurodegenerative diseases: the Ab peptide of Alzheimer's disease, the ABri peptide of Familial British dementia, a-synuclein of Parkinson's disease, the human prion and Doppel proteins of Mad Cow Disease, and the amyloid precursor protein of Alzheimer's disease. Some of the projects are summarized below:
Amyloid Aβ Peptide of Alzheimer's Disease.
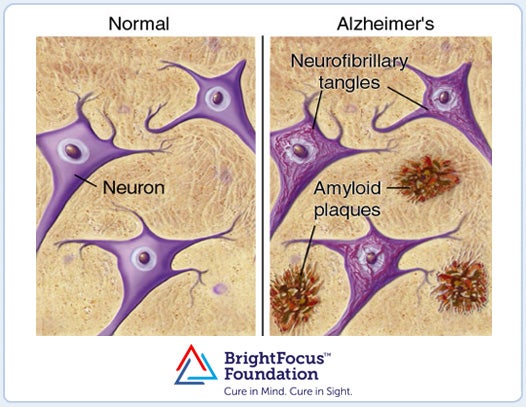
The brains of patients with Alzheimer's disease are characterized by the abundance of extracellular amyloid plaques and intracellular neurofibrillary tangles. The major constituent of the amyloid plaques is the Aβ peptide that contains either 40- or 42-amino acid residues (Aβ(1-40) or Aβ(1-42)). Their pathologic effects are related to their propensity to form insoluble aggregates dominated by β-sheet structures.
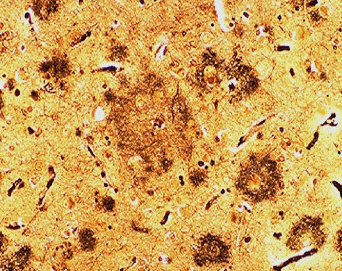
Figure 1. (A) Illustration of tissues taken from a normal and AD brain, and (B) picture of an actual amyloid plaque as seen under a microscope.
The mechanisms by which the Aβ peptide forms amyloid deposits remains unknown on a molecular level. To gain insight into this area, the Zagorski group has been systematically examining the structures and stabilities of synthetic Aβ peptides as a function of pH, solvent polarity, and in the presence of a membrane-like surface. This approach revealed that, depending upon the conditions, the Aβ peptide can adopt different conformations and aggregation states in solution, and that an α-helix-to-β-sheet conversion may be involved in amyloid production. The structures produced in these studies will guide in the development of inhibitors to prevent amyloid formation in Alzheimer's disease.
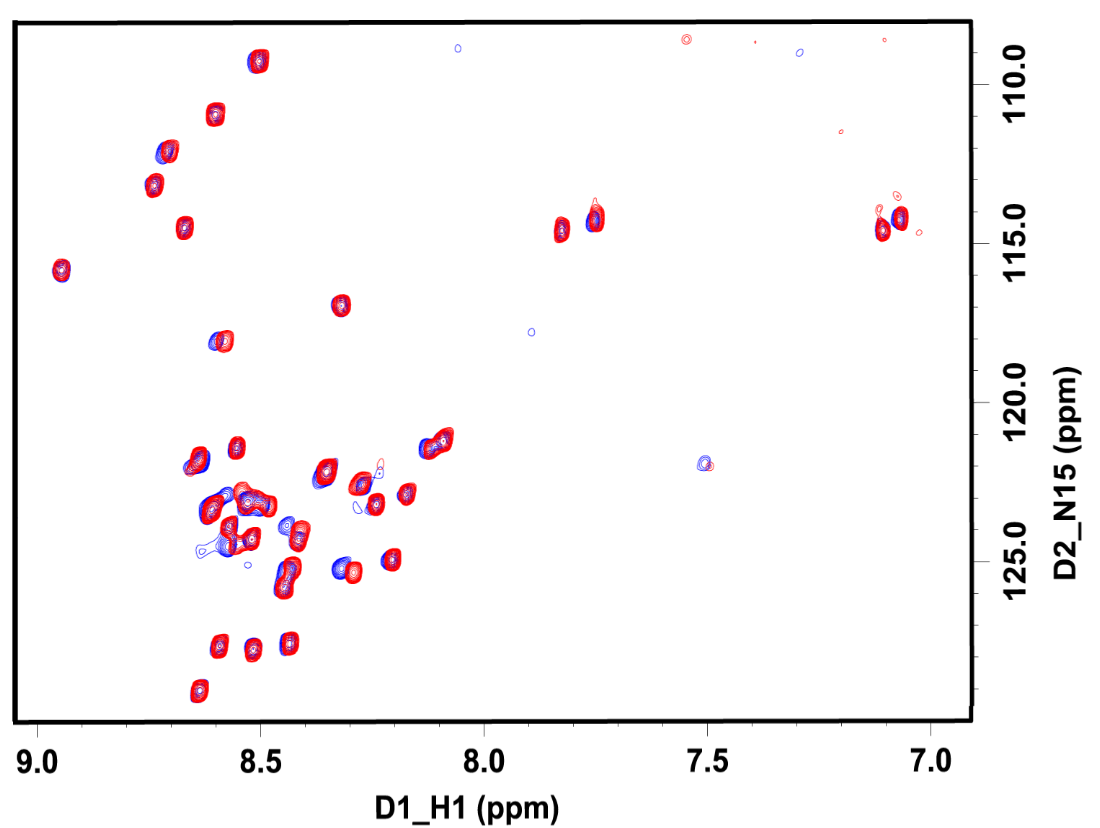
An ongoing project involves using NMR to characterize the structure of early formed aggregates, commonly known as the Ab-Derived Diffusible Ligands (ADDLs). Current thinking is that these early aggregated forms are the real culprits responsible for the Ab induced neurotoxicity rather than the later stage amyloid plaques. Representative HSQC NMR spectra of uniformly 15N enriched Ab(1-42) ADDLs (before and after centrifugation) is shown in Figure 2. The HSQC spectra show signals for every hydrogen atom that is directly bonded to a 15N atom.
Figure 2. Overlaid 1H-detect 15N HSQC spectra taken for the ADDL sample preparation, before centrifugation (blue) and the supernatant after the centrifugation (red). The residues showing chemical shift perturbation are located at the N-terminal half of the Aβ(1-42) (Glu3-Leu17).
α-Synuclein Protein.
The α-synuclein protein produces amyloid-like deposits in Lewy bodies, which are fibrous cytoplasmic inclusions characteristic of nigral dopaminergic neurons in the Lewy body variant of Alzheimer's disease and Parkinson's disease. Knowledge of the molecular mechanisms and the structure of α-synuclein is absolutely essential for the development of therapeutic strategies to treat these disorders. The major goal of our work is to determine the soluble structure of α-synuclein using NMR and mass spectrometry. The tertiary structures of alpha-synuclein when bound to the inhibitors is being explored by NMR methods.
ABri Protein of Familial British Dementia.
The ABri is a 34-residue peptide that is the major component of amyloid deposits in Familial British dementia, which is a human disorder that is autosomal dominant and characterized by dementia and cerebellar ataxia. In the amyloid deposits, the ABri peptide adopts aggregated β-pleated sheet structures, similar to those formed by the Aβ peptide of Alzheimer's disease and other amyloid forming peptides. As a first step toward elucidating the molecular mechanisms involved in FBD amyloidosis, we are exploring the effects of environmental variables (pH, buffer, peptide concentration) on the rate of β-sheet production. Using circular dichroism, atomic force microscopy, and electron microscopy, we have found that the ABri solution structure is strongly influenced by pH and concentration. Together, these data suggest that environmental variables are important variables in encouraging amyloid formation by the ABri peptide, consistent with the behavior of other amyloid forming proteins.
Human Prion and Doppel Proteins.
The prion diseases consititute a variety of disorders affecting the central nervous system of animals and humans. The first prion disease (scrapie) was discovered in sheep and cattle, and later found in humans as Gerstmann-Sträussler-Scheinker disease, Creutzfeldt-Jakob disease and fatal familial insomnia. The human diseases occur in sporadic, transmissible, and inherited forms, with the inherited/familial forms caused by mutations in a gene producing the prion protein (PrP). According to the protein-only hypothesis, an abnormal (scrapie-Sc) form of the prion protein (PrPSc) can induce the normal cellular counterpart (PrPC) to change structure (PrPC to PrPSc), which in turn can itself act as a transmissible agent causing further conversions of PrPC. Thus, the prion proteins can propogate and convert normal proteins into pathogenic forms by simply inducing a change in the molecular structure. When the accumulation of PrPSc reaches a critical mass, the observed pathology develops, which includes neuronal loss, astrocytic gliosis and spongiform changes. Our projects involve extensive collaborations with Drs. Witold Surewicz and Chen in the School of Medicine and we propose that many of the mutations are pathogenic due to alterations in the structure and/or folding properties of the PrP and Doppel proteins. To test this hypothesis, we are determining the NMR tertiary structures for several mutant PrP and Doppel proteins.
|