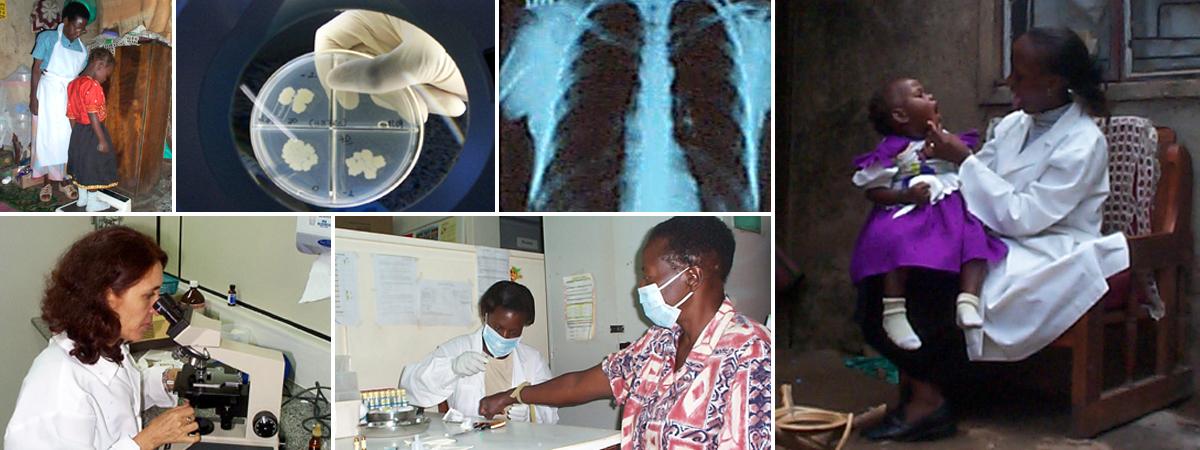
Working internationally to identify new approaches to prevention, diagnosis, and treatment of TB in areas of the world where it is most common.

Highlighting a 30 Year Partnership with Uganda
CWRU’s 30-year partnership with Uganda has earned worldwide recognition in research, training and capacity building
in HIV/AIDS and TB.

Our Research
The overall goal of the clinical studies to be carried out by the TBRU is to fill critical gaps in translational TB research and to provide tools needed to advance new health care interventions in TB endemic countries.